ab initio Modeling of Ethane - Hydroxyl Radical Reaction
Background
Ethane molecules in the atmospheric or in combustion reactions are oxidized to carbon dioxide and water via a series of reactions that is initiated via the reaction of ethane with an hydroxyl radical:
C2H6
+ OH. --> C2H5. + H2O
Computational Aspects
A series of ab initio calculations were made to predict classical exoergicity (no zero point energies included) of the ethane-hydroxyl radical reaction. HF, MP2 and DFT geometry optimizations and energy calculations, using four levels of basis sets for each of the three methods, were made for each of the four reactants/products, giving the following results:
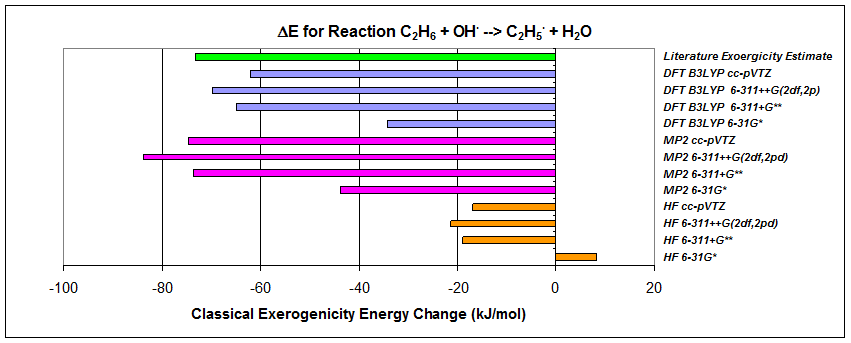
|
Basis set / Method |
HF |
MP2 |
DFT B3LYP |
|
6-31G* |
8.236 |
-43.77 |
-34.414 |
|
6-311+G** |
-18.881 |
-73.744 |
-64.973 |
|
6-311++G(2df,2pd) |
-21.347 |
-83.724 |
-69.864 |
|
cc-pVTZ |
-16.804 |
-74.718 |
-62.027 |
Table of
calculated exoergicities (vs literature estimate of -71 to -75 kJ/mol)
for ethane hydrogen abstraction via the hydroxyl radical
(literature estimate reported in Chuang, Y; Coitino,
E; and Truhlar*, D.G., J. Phys. Chem. A,
2000, 104, 446-450)
Findings:
- Correlated methods were essential for exoergicity results. HF methods performed poorly.
- Extended MP2 and DFT basis calculations gave results within several kJ/mol of current literature best estimates for this reaction